
近日,物理与电子信息工程学院耿文通教授团队在国际著名期刊Nature Protocols上发表题为Empowering materials science with VASPKIT: a toolkit for enhanced simulation and analysis的重要研究论文。
计算材料科学融合物理、材料科学和计算机技术,基于物理学和材料科学理论,借助高性能计算,实现从原子到宏观尺度的多层次模拟。基于密度泛函理论的第一性原理计算是一种从电子结构层面研究材料性质的方法,它不依赖经验参数,仅从量子力学原理出发求解电子结构,可准确描述材料的力学、热学、电学、光学和磁学等性质,已成为计算材料科学的核心方法。在众多的第一性原理计算软件中,利用Vienna Ab initio Simulation Package(VASP)最多。
耿文通团队自主开发了高通量材料物性计算与分析工具VASPKIT,该工具是一款面向第一原理计算用户的高效前后处理工具包,广泛用于晶体结构、电子结构、声子特性等的分析与处理。自2013年发布以来,已被全球科研工作者广泛采用,累计下载量超过18万次,每月下载量约3000余次。VASPKIT 软件的算法论文于2021年发表在Computer Physics Communications(2021, 267, 108033),4 年内被SCI论文引用4300余次(谷歌4900余次)。在2021 年至今全球所有研究领域已发表SCI 论文中,其引用次数排名第28,在已发表的所有中国科研单位参与的所有研究领域SCI 论文中引用排名第4,在全球已发表的物理领域SCI论文引用排第2。
近期,耿文通团队对VASPKIT在自动化程度和数据处理效率方面进行了大幅度的提升,相关成果发表在Nature Protocols。论文详细介绍了VASPKIT的新功能与使用方法,并通过图示范例展示其在弹性常数、能带结构与态密度计算中的高效表现等。
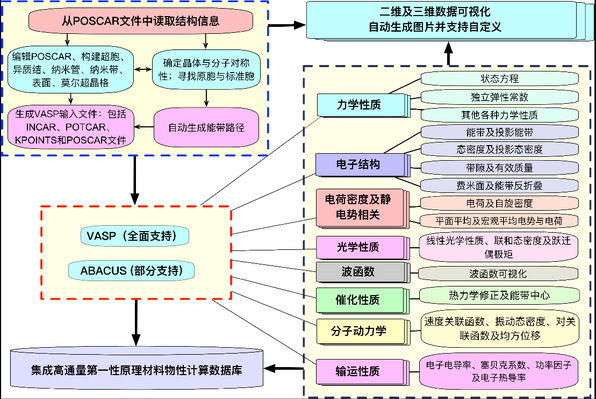
VASPKIT软件的架构图
VASPKIT的应用领域除物理、化学、材料与冶金外,还拓展至水资源学、食品科学、肿瘤学、遗传学、神经科学、农业、矿物学、环境生态学等诸多交叉学科。耿文通团队建立了全球开放的VASPKIT 软件中文网站和英文网站,及时发布软件的最新更新与优化信息。为促进研究人员的高效使用与学术交流,设立了学术公众号VASPKIT(目前关注人数超8000),并针对用户留言关切的特定功能,不定期发布详尽的使用指南。同时,构建了活跃的社群生态,组建了两个QQ 交流群,成员总数超3000,并设立了学术论坛,为研究人员提供便捷、多元化的互动交流平台。
Nature Protocols作为Nature子刊之一,创刊于2006年,覆盖物理学、化学、生物学等众多领域,专注报道重要研究方法和研究技术。
编辑:盛灿灿
